Easy Guidance for Abbreviated, Special, and Traditional 510(k) Clearance Applications

With medical device technologies evolving at an unprecedented rate, the FDA has experienced an overload of 510(k) submissions. In September 2019, the FDA revised its guidance documents for this submission in order to make it more streamlined and keep up with market demands.
The Premarket 510(k) submission is the main pathway for medical devices to receive market approval here in the US. With constant updates on existing regulations, it can get confusing to figure out the best regulatory method for your device. The following guide will clarify the types of 510(k) submissions there are giving your team a great starting point for regulatory submission.
What is Substantial Equivalence?
"Substantial Equivalence" is the method used by the FDA to prove to them that your new device is at least as safe and effective as the predicate device.
A device is substantially equivalent if, in comparison to a predicate, it:
- has the same intended use as the predicate
- has the same technological characteristics as the predicate
- has different technological characteristics and the information submitted to FDA
- does not raise new questions about safety and effectiveness
This does not mean that the devices must be identical, as there are a number of factors that go come into play when establishing equivalence such as design, materials, manufacturing process, performance, safety, effectiveness, labeling, etc.
Premarket Notification 510(k) Pathways
Using the guidance documents, we have outlined the FDA's three established timelines for reviewing a 510(k) submission:
Traditional 510(k):
- Used by most companies introducing a new Class II device and must submit a Traditional 510k Premarket Notification (21-section document).
- FDA’s clearance of a Traditional 510k is 90 days.
Special 510(k):
- Used when there is a design change or a modification to a device you have previously cleared that does not affect the intended use.
- FDA’s clearance of a Special 510k is 30 days.
Abbreviated 510(k):
- Used when there is guidance documents from the FDA and a special control has been established by the FDA for this device and consensus standards.
- FDA's clearance of Abbreviated 510(k) is 90 days.
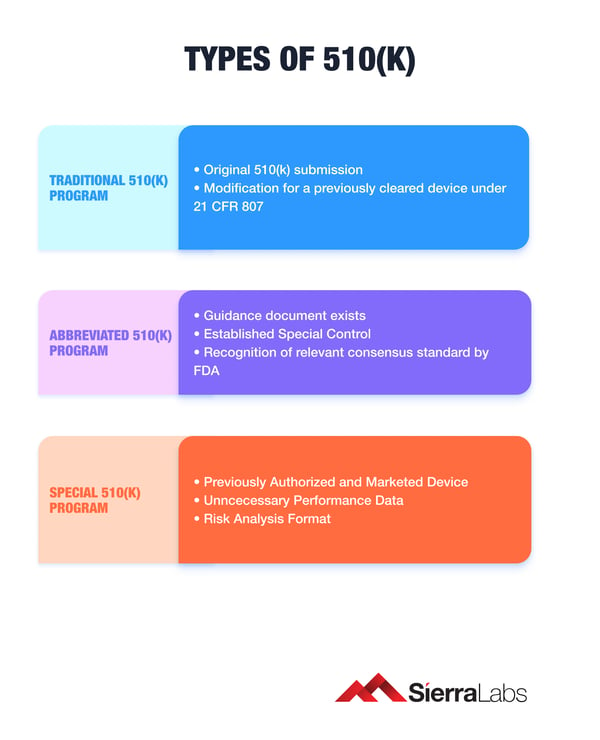
Traditional 510(k) Program
Is the basic pathway for any medical device regardless of its class type. The 510(k) premarket notification program is primarily intended for medical devices that are “substantially equivalent” (using 21 CFR 807) to other medical devices that have previously received market clearance from the FDA.
Manufacturers may use the traditional format when:
- PMA Not Required
- Exclusion from this type of submission applies to those devices for which Premarket Approval Application (PMA) is required.
- Exclusion from this type of submission applies to those devices for which Premarket Approval Application (PMA) is required.
- Established SE
- Provide sufficient information to allow FDA reviewers to determine whether the subject medical device is substantially equivalent to an identified predicate device.
Used by the FDA as an optional approach to the Traditional 510(k) Program to demonstrate SE in premarket notifications. Device manufacturers can choose the Abbreviated 510(k) pathway to demonstrate SE in premarket notifications by using one or more of the following:
- FDA Guidance Document
- Specifying device description, device design, risk management, and test methods used to address performance characteristics will help verify SE. Reports should also include information regarding any deviations or alternative methods used to demonstrate substantial equivalence that are not described in the guidance.
- Specifying device description, device design, risk management, and test methods used to address performance characteristics will help verify SE. Reports should also include information regarding any deviations or alternative methods used to demonstrate substantial equivalence that are not described in the guidance.
- Special Controls
- Specifying how this special control has been used to address a special risk or issue will help demonstrate SE. Special controls are defined in section 513(a)(1)(B) of the FD&C Act as those controls, such as performance standards, post-marketing surveillance, patient registries, development and dissemination of guidelines, recommendations, and other appropriate actions that provide reasonable assurance of device’s safety and effectiveness.
- Specifying how this special control has been used to address a special risk or issue will help demonstrate SE. Special controls are defined in section 513(a)(1)(B) of the FD&C Act as those controls, such as performance standards, post-marketing surveillance, patient registries, development and dissemination of guidelines, recommendations, and other appropriate actions that provide reasonable assurance of device’s safety and effectiveness.
- Voluntary Consensus Standard(s)
- Specifying the basis of use with supportive information and data must match the manufacturer's voluntary consensus standard that is recognized by the FDA.
Special 510(k) Program
The Special 510(k) pathway was previously limited to the review of changes to a cleared device that did not impact its intended use or alter the device’s fundamental scientific technology.
In the 2019 published version of the guidance, the FDA has revamped its approach to make this pathway more inclusive by allowing for specific intended use changes and technological changes to be reviewed under a Special 510(k).
Manufacturers can submit their device modifications as a Special 510(k) if they meet the following minimum requirements:
- Previously Authorized and Marketed Device
- The proposed change is submitted by the manufacturer legally authorized to market the existing device, meaning the modification must be made to the manufacturer’s own device.
- Unnecessary Performance Data
- Performance data is unnecessary or may be evaluated using well-established methods which are available by the FDA to evaluate the change.
- Risk Analysis Format
- All performance data necessary to support substantial equivalence (SE) can be reviewed in a summary or risk analysis format.
Convert A Special 510(k) to a Traditional 510(k)
FDA can convert the Special 510(k) to a Traditional 510(k) if SE determination cannot be made through information submitted in a Special 510(k). If there is an issue, the FDA will include this in the Refuse To Accept (RTA) letter.
This type of conversion results in delays as the content of the Traditional 510(k) varies from a Special 510(k) and this data gap needs to be fixed for a complete review. Therefore, in cases where the manufacturer is unsure about use of Special 510(k) pathway, a Pre-Submission meeting with FDA can be explored further to achieve regulatory success.
Defining a Regulatory Strategy
Since every submission is unique in the eyes of the FDA, some manufacturers might have a tougher time finding "substantial equivalence" or a predicate for their device. This will in turn cause a delay in submission. Along with guidance updates, the FDA has become more flexible in engaging with newer medical device companies to answer any questions or concerns that may arise.
This engagement process is all part of your regulatory strategy, which is something we advise you to have even before submitting any documents! If you need some further insight to how a regulatory strategy works, be sure to check out our 8 Guidelines for Your Medical Device Regulatory Strategy.
Benefits of Utilizing Pre-Submissions
Even with an action plan, it can be easy to miss important steps throughout the process of your regulatory submission and we want to assure you avoid any delays in time and organization. Pre-Submissions can be a beneficial way to effectively prepare for your 510(k) submission to FDA.
FDA Pre-Submission program is a meeting with the FDA where they provide you with feedback before submitting your formal medical device application. It is voluntary, not mandatory, and is a way for you to fix gaps in your application before submitting your final version. A sort of practice exam if you will, with detailed feedback on ways to improve for the real deal.
We have helped various medical device companies like yours prepare for 510(k) submissions in each of the varying timelines. We have also led them through Pre-Submission programs, assuring they are comfortable and confident in their FDA interactions.
We know speaking with the FDA can be a daunting first step toward achieving market approval. For your convenience, we have created a free questionnaire to help you prepare for any questions the FDA may have. Try it below to help you get ready for your meeting with the FDA!
Want to see how Sierra QMS can help your organization?
Click Here for a Free Trial!

Quick and easy!