Learn how your company can form a Regulatory Strategy in 8 easy steps.

In this blog, we will explore:
How to Develop A Regulatory Strategy
In today’s competitive marketplace, it has become more and more difficult for medical devices to stay compliant under the FDA’s regulations. Because of this, your product team needs a regulatory strategy with established project deliverables and resources required before talking to the FDA.
A regulatory strategy means constructing a documented plan that describes the specific steps and actions required to successfully stay compliant. It also contains the elements needed for a regulatory submission.
Before reading over the guidelines, your team must first identify a couple of characteristics about your product, such as determining if it is technically a medical device. The team will list everything known about the product, such as:
- What is the device?
- How is it to be used?
- What markets will be served?
- Is this a new/novel product or is it a modification of an existing device?
- Are there similar devices being distributed?
- What claims will be made?
Answering these questions will help you dictate your device’s journey to the market.
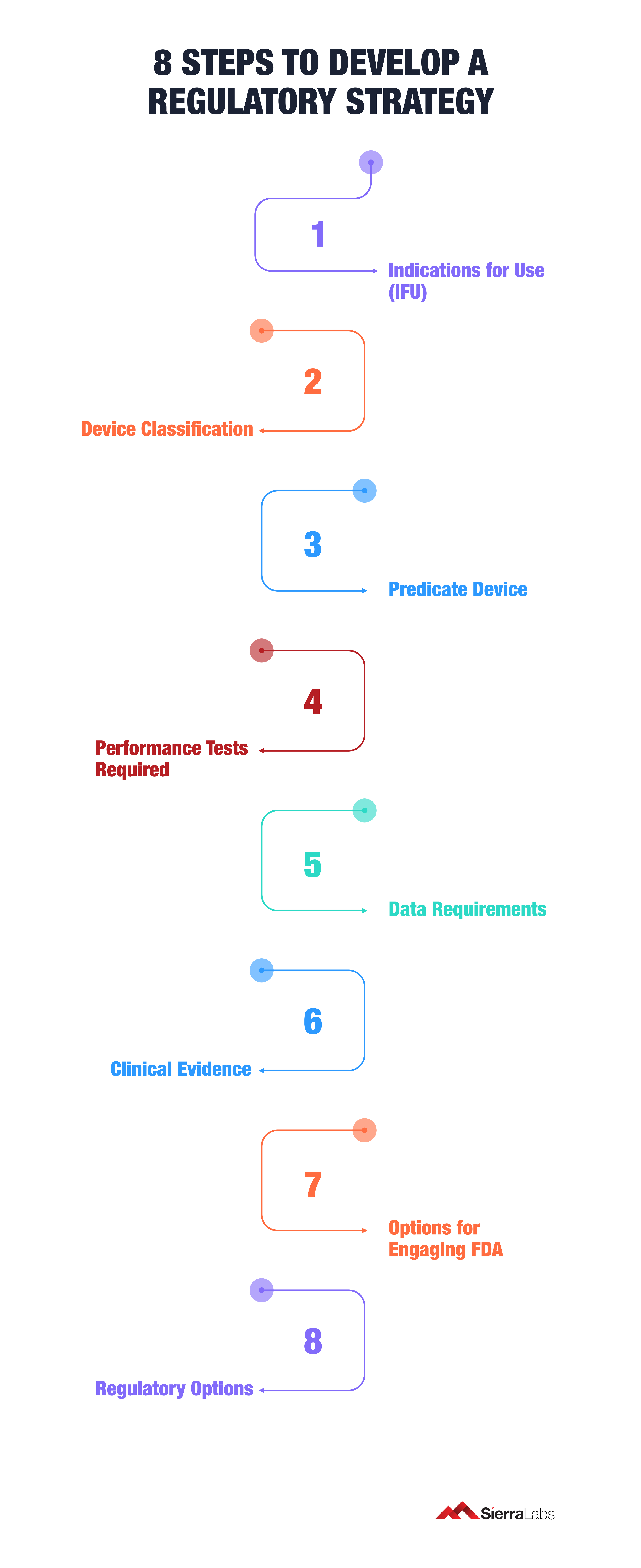
Here are the guidelines:
1. Indications for Use (IFU)
- Your team should develop an IFU (a basic description of how the device is intended to be used), and should include:
- Diagnostic purposes on the safety and effectiveness (Ex. screening, real-time monitoring, etc.)
- Intended population (age, patient group, diagnose type)
- Intended setting or environment (ex. in home, under supervision of a healthcare professional, etc.)
- Any prohibitions on certain populations (due to differences in diversity, growth, or development milestones)
2. Device Classification
- Your team should find what Risk Classification category your device falls under:
- Class I: General Controls - With Exemptions/Without Exemptions
- Class II: General Controls and Special Controls - With Exemptions/Without Exemptions
- Class III: General Controls and Premarket Approval
3. Predicate Device
- Your team should identify the predicate device (also known as legally marketed device(s) with substantial equivalency). Substantial equivalence is established with respect to:
- The traded name under which the device is marketed
- Name of manufacturer(s) who developed the device
- Marketing status, i.e., pre-amendments or post-amendments device
- 510(k) numbers for post-amendments devices
- Classification information, i.e., product codes, classifying regulations, etc., for the device
4. Performance Tests Required
- Your team should gather all necessary design and manufacturing requirements such as:
- Chemical, physical, and biological properties
- Infection and microbial contamination
- Manufacturing and environmental properties
- Protection against mechanical risks
- Protection against the risks posed to the patient by supplied energy or substances
- Protection against the risks posed to the patient for devices for self-testing or self-administration.
5. Data Requirements
- Gather studies done to date existing data that could be leveraged during your meeting with the FDA. For areas in which no evidence exists, new studies will be required. The studies should include:
- Bench
- Animal
- Biocompatibility
- Software
- Electromagnetic compatibility
6. Clinical Evidence
- Record the overview of the intended timeline for clinical investigations to support the safety and effectiveness of the device. The main design elements to record include:
- Sampling frame
- Number of subjects
- Clinical setting
- Methods
- Outcomes
7. Options for Engaging FDA
- These are the ways your team can initiate a conversation with the FDA:
- 513(g) Request for Information
- In cases where you can’t find a predicate device, your team can complete a request for more information via this form. Your team must submit a description of the product, the proposed intended use and indications for use, and its proposed classification to the agency for review. An example of this looks like:
- "Does the FDA agree with our determination of Class II based on this, this, and this...?"
- In cases where you can’t find a predicate device, your team can complete a request for more information via this form. Your team must submit a description of the product, the proposed intended use and indications for use, and its proposed classification to the agency for review. An example of this looks like:
- Pre-submission Feedback
- The FDA will then respond within 60 days addressing whether the product is considered a medical device, and if so, stating its classification and whether it will be subject to active regulation or enforcement discretion.
- If your team is unsure about choosing the right regulatory pathway for your device, then a Pre-Submission might be the choice for you. The pre-submission process facilitates a dialogue between you and the FDA
- To do this you must compile a package of information on the device such as proposed indications for use statement, details of the testing conducted to date, a full protocol for planned preclinical and clinical studies, a summary of the proposed regulatory pathway, questions for which the company is seeking feedback, and other required administrative information.
- 513(g) Request for Information
8. Regulatory Options
- Determine the Regulatory Options for your medical device with key data considerations based upon the information gathered above.
- Be sure to include any complications or successes about the regulatory strategies of competitive devices to help anticipate how prior or current requirements might evolve over the product development timeline.
How to Implement your Regulatory Strategy
After following these guidelines, your team will be on track to designing a comprehensive regulatory plan for your product’s development. This plan will contain key project deliverables and proper resource requirements for your team to assess a timeline.
As you can guess, this regulatory plan will demonstrate a roadmap to the FDA as it contains the main regulatory elements, such as: comparisons to predicate devices, supporting data, specific pre-clinical and clinical reports, and information from pre-submission meetings.
A solid regulatory strategy is one of the foundations of successful medical product development. Start by asking a broad range of questions to ensure there is a solid understanding of the product so that there aren't any unprecedented regulatory impacts.
You are not alone…
As previously stated above, filling out a Pre-Submission is a great way to get a head-start in engaging with the FDA. The guidelines above will help you gather all the right information before having a formal meeting with the FDA.
Even with these guidelines and other online resources, the Pre-Submission process can still be a fairly unclear and stressful process. You do not want to waste your precious time with FDA or risk not gaining FDA approval your first time around, and there are a lot of materials to prepare.
We are here to help your team design a strong and efficient regulatory strategy for your medical device! Sierra Labs has guided various organizations in navigating the complex regulatory environment, while simultaneously propelling their medical devices to market.
We have also developed a free questionnaire that allows you and your team to determine if you’re ready for FDA's Pre-Submission program! Try it below to help you get ready for your meeting with the FDA.
Are you ready for FDA Pre-Submission?
Try our Free Questionnaire!

Quick and easy!