How embracing regulations can give you a leg up on your competition.

If your company conforms to ISO 13485, does it really matter if you are missing any of the regulatory requirements?
The short answer, is yes. The primary focus in the latest edition of ISO 13485 is regulatory compliance and in reality, an organization cannot conform to ISO 13485 in its true sense unless the applicable regulatory requirements are met first. Thus, it is important for both medical startups and established companies to understand these regulatory requirements, which are there in the market to protect public health interest. Most people view these regulations as a hectic burden, however, they are for the mutual interest of all parties within the market and, if embraced effectively, can be your competitive advantage!
ISO 13485:2016 standard keeps both regulators and customers in one group, from the requirements for product realization and feedback, to measurement, analysis, and improvement. Moreover, the management responsibility function of the Quality management system also interacts with both regulators and customers as shown in the illustration below.
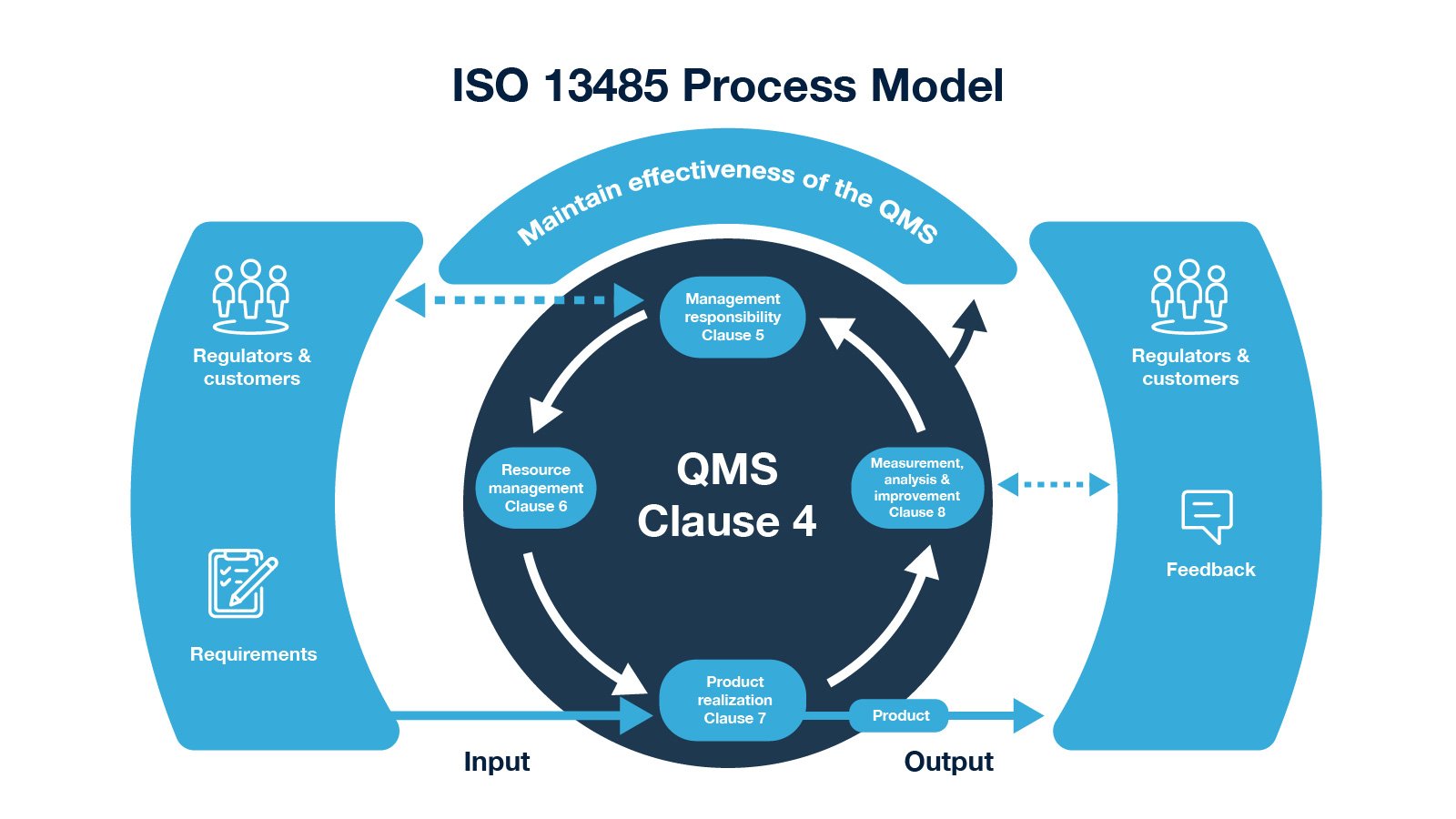
Fig. 1: ISO 13485 Process Model from ISO 13485 vs. CFR Part 820 blog.
For a detailed breakdown of ISO 13485, read "What is ISO 13485:2016?" before continuing!
The Source of Regulatory Requirements
The standard clarifies that regulatory requirements are applicable laws, regulations, ordinances, and/or directives. ISO 13485 standard treats regulatory requirements in the same essence as the customer requirements, because to reach the customer in a medical device industry, regulations are imperative. The standard directs the companies to identify which regulatory requirements are applicable to its Quality Management System (QMS) and their impact on the QMS.
Regulatory bodies lay down the requirements regarding numerous aspects of the medical industry, focusing on product safety, service provision, consumption of raw materials, customer communication as well as communication with regulatory authorities, and other manufacturing operations. Non-compliance, whether deliberate or not, can result in grave consequences ranging from expensive fines, to a ban on marketing products, etc. One needs to identify the regulatory authorities in the countries where its products or services will be marketed and be in full compliance.
Understanding Entities:
Regulatory authorities: Every country has a regulatory body that regulates the industry of medical devices.
- Examples: FDA, EMA, etc.
Directives or Acts: In few countries, Directives or Acts regulate the supply of medical devices and related services.
- Examples:
- Council Directive 93/42/EEC is the European Commission’s Medical Device Directive, and it provides requirements related to needed durability and quality performance in medical devices.
- Regulations such as FDA 21 CFR Part 820 in the U.S. are also there in place of a Directive or Act.
Statutory instruments: Few countries have statutory instruments managing the medical device industry.
- Example: Statutory instrument No. 252/1994, European Communities (Medical Devices) in Ireland, regulates producers and providers of medical devices to comply with requirements given within that.
What is the relation between regulatory requirements and ISO 13485?
According to ISO 13485, an organization has to meet or exceed all applicable legal requirements. An organization cannot be compliant with the standard if it does not fulfill regulatory requirements. ISO 13485:2016 incorporates some specific requirements that deal with the knowledge you should have acquired while determining relevant regulatory requirements. After all, you will have to comprehend such requirements so as to manage QMS processes. ISO 13485, guidance on managing regulatory requirements, include:

Fig. 2: Guidance clauses for managing regulatory requirements.
FDA Medical Device Classification Process
There are different classes for medical devices when entering the market; these classifications are made by regulatory bodies in order to manage the industry efficiently. Every country has its own set of classifications.
A simple medical device, such as surgical scissors, may not require clinical data and additional control, such as a lifesaving incubator would. Therefore, regulators apply controls where needed and do not unnecessarily burden the suppliers and manufacturers. In the case of the US market, FDA has made three different classes for medical devices. Each class is regulated through general controls, then Class II and Class III through special controls, and finally Class III requires additional clinical data and Premarket Approval Application (PMA) process as shown in the illustration below.
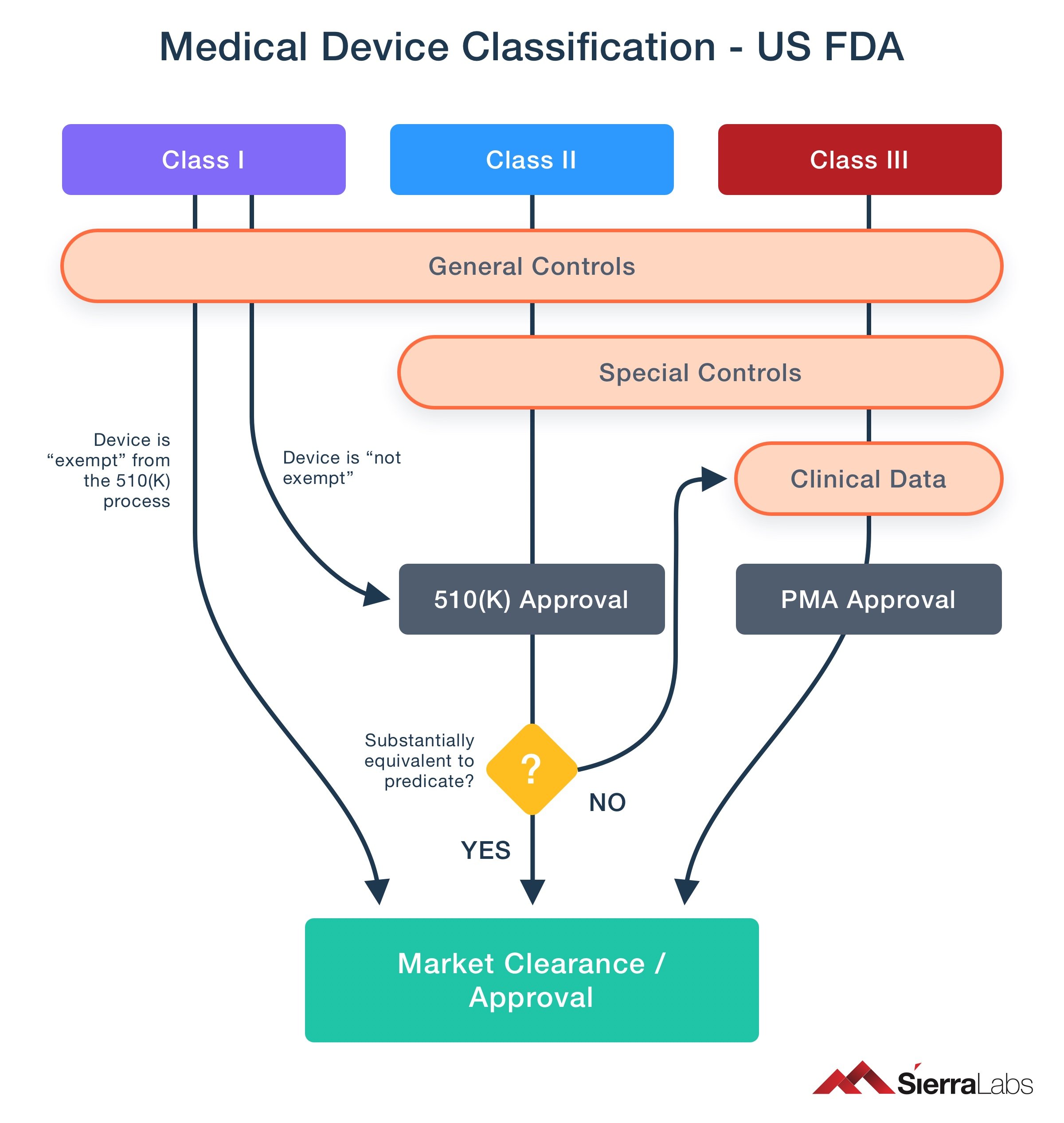
Fig. 3: Medical Device classification process.
Your QMS needs to incorporate applicable regulatory requirements!
One of the important steps in ISO 13485 implementation, and becoming more vigorous in approaching the market, is complying with regulatory requirements. With compliance to regulatory requirements, an organization will be prepared to offer products which are safe, and avoid the setbacks (and disadvantages) related with noncompliance.
The optimized solution to reduce risk, maintain quality, and accelerate innovation is by utilizing an FDA compliant and best practice conformant medical device quality management application. Sierra Quality Management System (QMS) offers you a robust start to identify relevant regulatory requirements, and to evaluate your company’s level of compliance.
Sierra QMS is designed for organizations who are looking to market medical devices in a global regulated environment. It is built for engineering teams to operate with their preferred tool-sets while automating the compliance with medical device QMS principles for global markets. Sierra Labs helps both medical device software developers and SaMDs developers to build a vigorous QMS that includes a variety of effective workflow management tools.
Want to see how Sierra QMS can help your organization achieve Conformity AND Compliance?
Download our free White Paper to learn more!
