A comparative study on the key differences between FDA’s QSR and ISO’s QMS.
Comparison: FDA 21 CFR Part 820 vs. ISO 13485:2016
ISO 13485:2016 is an international standard for Quality Management Systems of medical device manufacturers and providers. The U.S. standardization body, ANSI (The American National Standards Institute) has been involved in the revision of ISO 13485 standard in 2016, and served as the secretariat for the revision work.
FDA Regulators now intend to increase attention to medical device safety, reinforce effective risk management, and enhance reporting to the authorities. The new version of ISO 13485 is expected to support these focus areas.
Major portions of Part 820 regulation requirements are incorporated in ISO 13485:2016. But, there are a few minor specifications that might not be explicitly contained within ISO 13485. The FDA is presently working on the proposed rule, which will be issued by 2019, that can make the transition to ISO 13485 possible in the US for regulatory purposes. The rule is titled "Harmonizing and Modernizing Regulation of Medical Device Quality Systems".
Why the FDA is Shifting towards ISO 13485
The intent is to coalesce the FDA’s Quality System Regulation (QSR) with ISO 13485:2016 in an attempt to harmonize requirements while revamping the regulations, which hasn’t been updated in more than 20 years. Medical device companies have had to comply with both requirements, as well as other international requirements. Since international medical device regulators (including the ones in EU, Canada, Australia, and Japan) stand their quality system requirements on ISO 13485:2016, FDA can renew the regulation to align with a wider, universally trusted quality system standard.
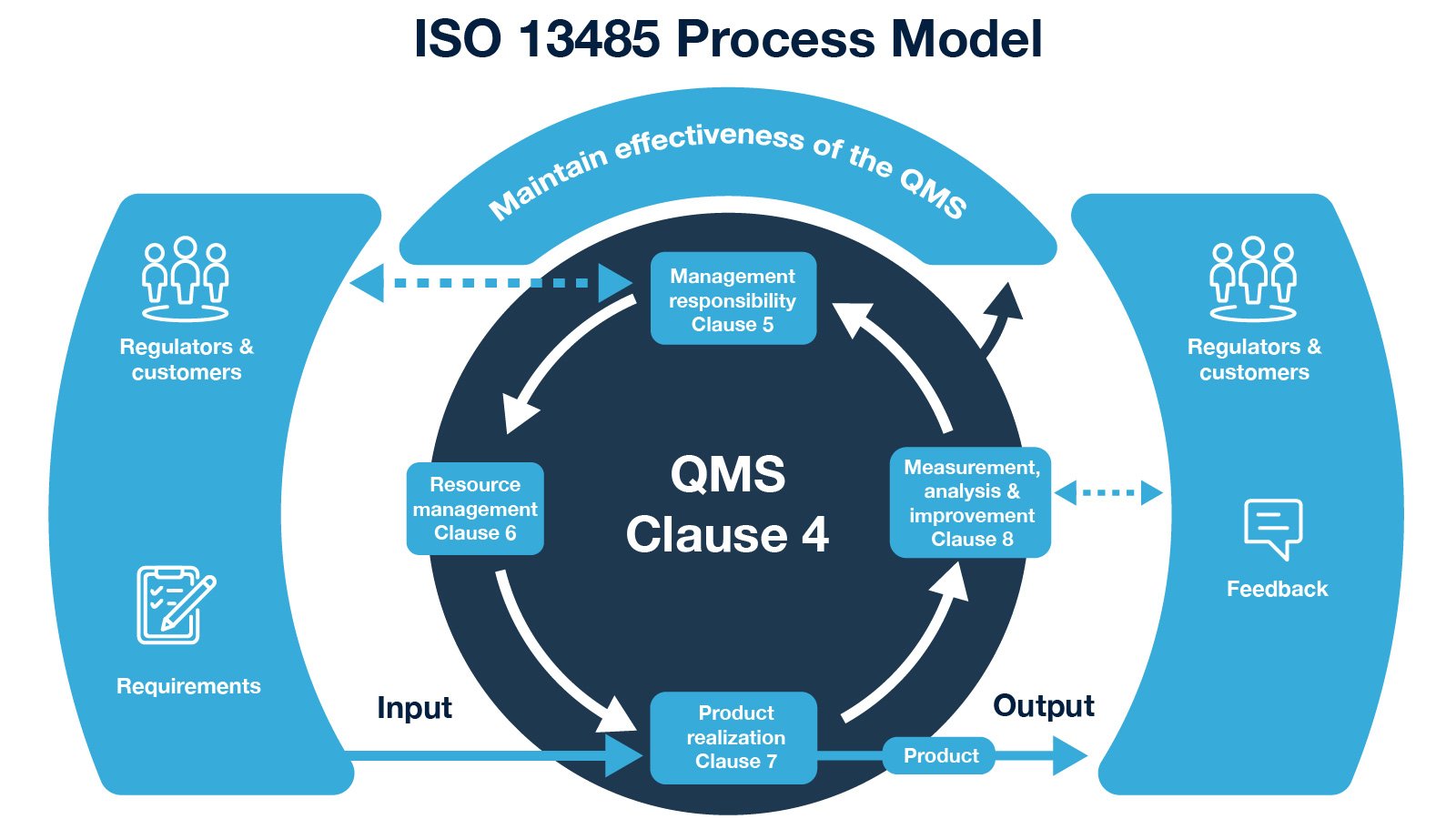
Organizations conforming to ISO 13485 standard can move confidently towards compliance of FDA 21 Code of Federal Regulation (CFR) Part 820 (Quality System Regulation). With compliance to the subject regulation, companies can market medical devices in the U.S.A commercially. Part 820 specifies requirements for the quality system to comply with, calling them current good manufacturing practices.
Part 820 is greatly aligned with ISO 13485 in the perspective of QMS requirements. There are other Parts of FDA requirements as well, for example:
- Part 810 – Related to medical device recall.
- Part 830 – Related to unique device identification of medical devices.
FDA 21 CFR Part 820 and ISO 13485 are correlated due to their application, scopes, and impacts on each other. The correlation matrix below will help you to appreciate the relationship and differences between quality standard and regulation, application scopes, and more.
| ISO 13485:2016 (QMS) | FDA 21 CFR Part 820 (QSR) |
| 13485:2016 prioritizes a risk-based approach and cites related standards, for example, ISO 14971 for managing medical device product risk management. | FDA Part 820 rarely specifies requirements for risk management. |
| ISO 13485 is an internationally accepted standard that offers an approach to abide by common regulatory requirements. | Other countries have their own regulations; FDA 21 CFR Part 820 is solely a U.S. regulation. |
| ISO 13485 standards have undergone multiple revisions. | QSR structure remains unchanged since 1997. |
| ISO 13485 is standard, thus it does not mandate a compulsory documentation structure for a Quality Management System. However, there are various requirements that need to be addressed and the organization decides how to manage it. | The FDA does not require companies to follow a particular documentation system, however, all documentation needs have to be addressed. Nevertheless, companies working for the U.S. market themselves want to set up a documentation system aligned with Part 820 to ease their audit process. |
| When a requirement is met as per ISO 13485, it is known as conformance. | When a requirement is met as per FDA 21 CFR Part 820, it is known as compliance. |
| Conformance, to ISO 13485 standard, is an internal effort of the organization to please customers and to build trust. | Compliance, with Part 820, is an external requirement by the government in the U.S. market. |
| ISO 13485 is not a directive, regulation, or law, but rather an agreed international standard. | FDA 21 CFR Part 820 is a compulsory quality system regulation for the medical device market in America. |
To read 21 CFR Part 820 regulations and see how they interact with other standards/regulations, check out Sierra Policies!

The FDA’s Quality System Regulation Part 820 is more closely correlated with ISO 13485:2016 than its precedent ISO 13485:2003 and the commonly used QMS model of ISO 9001:2015. It is more suitable for companies to establish a homogenized Quality Management System to manage regulatory requirements in different countries.
A QMS that is Ready for the Shift
Now that you have learned all about the transition from FDA 21 CFR Part 820 to ISO 13485, let’s talk about ways to incorporate a QMS into your organization that is ready for the shift!
The optimized solution to reduce risk, maintain quality, and accelerate innovation is by utilizing an FDA compliant and ISO conformant medical device quality management application. Sierra Quality Management System (QMS) allows you to have conformity AND compliance in your production process.
Sierra QMS is designed for organizations who are looking to market medical devices in a global regulated environment. It is built for engineering teams to operate with their preferred tool-sets while automating the compliance with medical device QMS principles for global markets.
Want to see how Sierra QMS can help your organization achieve Conformity AND Compliance?
Download our free White Paper to learn more!
