Easy Ways to Meet FDA Regulatory Requirements with Your Software Development Plan

Keeping a close eye on each regulatory requirement will save you costs in the long run. If you fail to integrate regulatory strategy into your development process, expect to lose time or even increase the risk of liability concerns.
Effective Planning for SaMD Development
Due to the nature of the regulatory environment for SaMD, manufacturers must pay close attention to which requirements must be met before receiving market approval. This means the development process includes a pre-planning phase where proper procedures are put into place to create a sufficient trial of data.
Preparation is key to a development strategy and usually includes documents verifying data associated with:
- Valid Clinical Association
- Process of making a close association to a current software application that impacts a relevant healthcare situation or medical condition. This can also be known as “scientific validity.”
- Analytical Validation
- Manufacturers are expected to provide "objective evidence" of sound software development processes. This includes effective software design for data processing. This type of validation is also needed to confirm the safety, performance, and effectiveness of the software application.
- Clinical Validation
- To gain market approval, manufacturers must demonstrate the "clinically meaningful" performance of a SaMD application. This can be shown with an application that has a measurably positive impact on the individual or public health, including clinical outcomes, diagnostics, prediction of risk, or prediction of treatment response.
- Clinical validation may be gathered through clinical studies, but FDA guidelines also provide support for:
- Existing data from relevant studies for the same use
- Existing data from related studies for a different use
- Generating new clinical data
By planning to gather data from these SaMD validation processes, your team will start on a solid regulatory path and reduce potential setbacks when meeting with the FDA. Integrating compliance into the SaMD development lifecycle starts with proper data collection and documentation, which we will discuss later in this blog.
Schedule Reviews for Your SaMD
A key component for your SaMD development plan should include review sessions detailing how your individual development processes are complying with standards and regulations. ISO 13485 requires management review at planned intervals and clear documentation of these sessions.
You can learn more about ISO 13485 here.
Review sessions can be conducted using an iterative approach. This includes correcting any potential regulatory setbacks with controls or real-time changes. With formal review embedded within the development plan, your team can demonstrate to the FDA that the data collection and documentation processes are fulfilling regulatory policies for SaMD.
Document Every Process of Your SaMD Development
Documentation is everything!
Especially when it comes to SaMD, it's crucial to get across to your team the proper documentation expectations throughout the entire development process. Having an individual in charge of the review sessions and quality assurance of software documentation will improve efficiency when meeting project demands. Developers are the best fit for this role since they are working most closely to the software application.
Don't have someone in charge of QA/RA? Sierra Labs can act as your Quality Function!
Click here to view our consulting services.
Here is an example of proper documentation done from the beginning to end of a project:
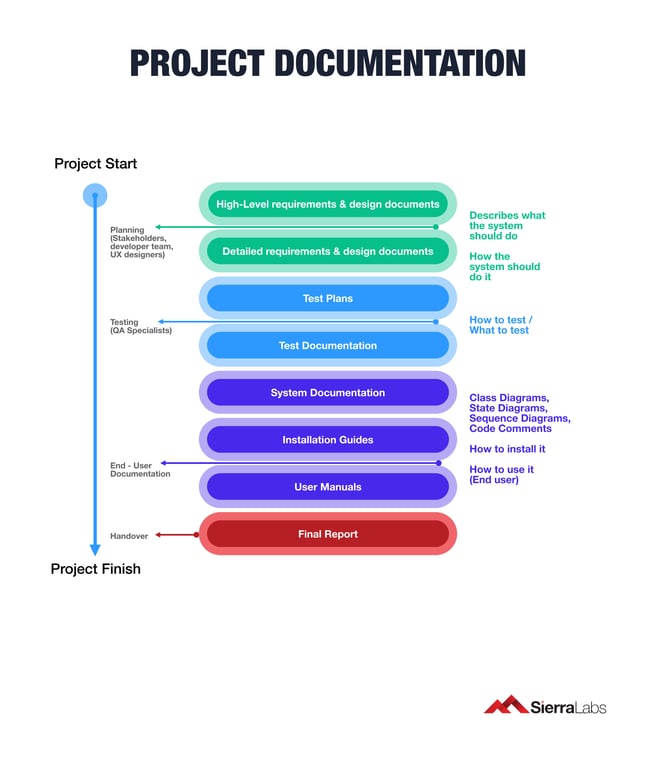
Instilling Efficient Management
Medical device manufacturers who are working to bring a SaMD product to market are subject to stricter regulatory requirements than manufacturers working on a traditional medical device. The requirements include rigorous guidance from the FDA on the real-time collection of data, constant product validation, and comprehensive documentation for each submission.
The right time to start thinking about regulatory requirements isn't after you've begun developing, it's during the product design phase before any code is written.
QMS for Medical Software Developing Companies
With a constant need to record every process, SaMD organizations must have an optimized solution for collaboration and document management. SaMD startups and scale-ups need a Quality Management System (QMS) that is built to scale with your growth, make documentation compliant, and meet regulatory standards like ISO 13485.
Sierra QMS is designed for organizations who are looking to market SaMD in a globally regulated environment. It is built for engineering teams to operate with their preferred tool sets while automating compliance with medical device QMS principles for global markets. Sierra Labs helps both medical device software developers and SaMDs developers to build a robust QMS that includes a variety of effective workflow management tools.
Want to see how Sierra QMS can help with your SaMD Development?
Download our free White Paper to learn more!
