Uncover which regulatory pathway is right for your medical device!

Why do I need a regulatory pathway?
Any device or product with even a minimal level of public risk must determine a regulatory pathway. If your company is creating a medical device or solution, chances are you will have to deal with the FDA.
Understanding what each path entails can help you designate a timeline for your teams to reach compliance. This can also minimize the amount of work and stress you put into the regulatory segments of your product. The various policies and regulations assigned by the FDA don’t have to become regulatory hurdles!
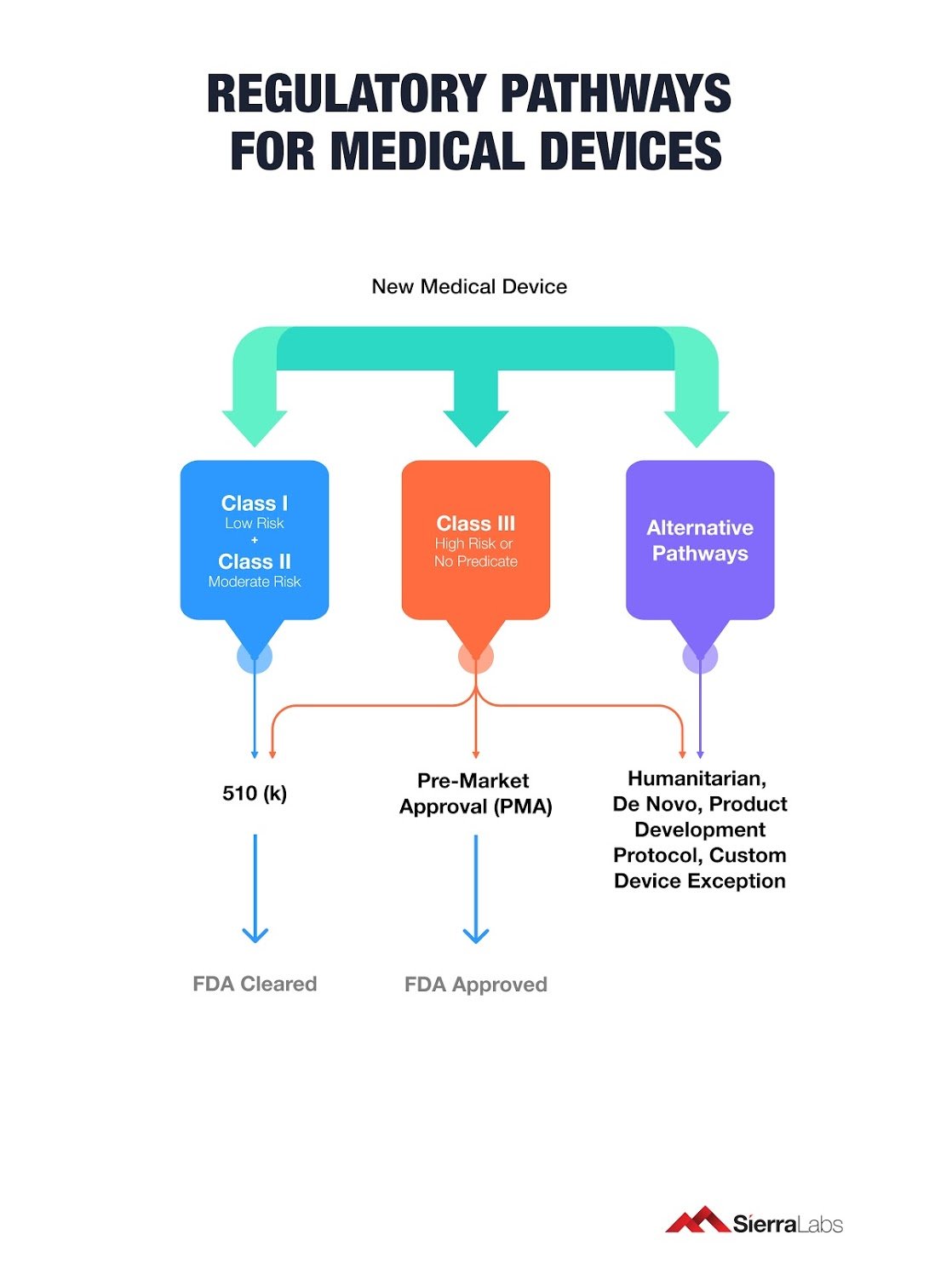
Here are the 6 Regulatory Pathways for Medical Devices:
- Pre-Market Notification 510(K)
- Pre-Market Approval (PMA)
- Product Development Protocol (PDP)
- De Novo
- Humanitarian Device Exception (HDE)
- Custom Device Exception (CDE)
Being aware of the various pathways can help you speed up time to meet your business goals. We are here to provide the proper information you need to choose a pathway that enables your medical device to reach the market as efficiently as possible!
Which Regulatory Pathway is Right for my Medical Device?
We’ve compiled all the information you need to know to choose the right path for your device! Let’s take a closer look at each of 6 regulatory pathways below:
Regulatory Pathways for Medical Devices
| Regulatory Pathway | Description | Steps |
Timeframe |
| 1. Pre-Market Notification 510(K) |
This is the most frequently used pathway by medical device companies with a Class I and Class II Risk Classifications. This pathway becomes more of a back and forth conversation with the FDA. The main goal in this pathway is to prove your medical device meets the standards and effectuality of a predicate device, one that is already on the market. |
(1) Compile information on your device’s design features and verification testing. (2) Contrast these features with the predicate device’s features. |
30-90 days |
| 2. Pre-Market Approval (PMA) | This pathway is for devices who are designated as Class III or High-Risk must use the Premarket Approval (PMA) pathway. |
(1) FDA staff will determine completeness through an administrative and limited scientific review. (2) FDA staff will conduct an in-depth scientific, regulatory, and Quality System review. (3) An advisory committee will review and offer any recommendations. (4) Any final deliberations will occur and the FDA will document and notify you of their final decision. |
180 days |
| 3. Product Development Protocol (PDP) | This pathway is an alternative for medical devices with a Class III or High-Risk Classification and is also technologically well-known in the industry. |
(1) Come to an early agreement or contract about the design and development activities undergone by the FDA. (2) Complete the agreed-upon milestones and report back to the FDA to receive a PDP. |
180 days |
| 4. De Novo |
This pathway is for devices with lower risk and does not qualify for 510(k) clearance, or cannot find an equivalent predicate in the market. A company that takes this pathway can potentially make their device a predicate and set a standard for future devices.
|
(1) Must present a robust risk mitigation strategy | Varies |
| 5. Humanitarian Device Exception (HDE) |
This pathway is meant for any device that is meant to treat or diagnose a select few who might have a rare condition or disease. This device must be a unique solution to treat or diagnose a condition or disease in order for the FDA to consider it an HDE and a predicate device. |
(1) Must obtain approval of exemption by the FDA. (2) Submit an HDE application to the appropriate review center. |
Varies |
| 6. Custom Device Exception (CDE) | Given to devices that are exempt from PMA or 510(k) submission, this pathway contains precise criteria due to its “customized” characteristics. |
(1) Must make proper modifications to the device to comply with the order of an individual physician or dentist, typically in the form of a prescription. |
Varies |
What comes next?
Now that you have gone over the 6 different regulatory pathways, you can start planning a proper regulatory strategy for your medical device. Choosing a pathway that matches your business’ expectations will have your company gain a better understanding of the time and resources needed to get your device to market.
There are resources and tools to make your medical device’s journey to market smooth and stress-free. Navigating the complexity of these various pathways can truly be made simpler, and all of the weight should not have to fall on your shoulders.
Here at Sierra Labs, we offer a Quality Management System (QMS) so that you and your business can robustly navigate through FDA regulations. Sierra QMS is designed for organizations that seek to market medical devices in a global regulated environment.
The optimized solution to reduce risk, maintain quality, and accelerate innovation is by utilizing an FDA compliant and best practice conformant medical device quality management application.
Need help planning your regulatory roadmap? We can help!
Download our free White Paper to learn more!

Ask us anything.